計算材料 2007 期末考 參考答案
一、
首先載入 GaN 結構,設定 Task 是能量,選擇 Property 是 Density of States,並且勾選 Projected Density of Sates (PDOS),發動一個計算。
計算完成後,開啟分析表單,選擇要做 PDOS,然後在晶胞視窗是開啟的情況下,點選其中一種原子(可按住 Ctrl 多重選取),按 View 顯示 PDOS,這就是所選原子(物種)的投影態密度。為了更加新晰起見,PDOS 選項中的 sum 與不會有的軌域(如 f)都可以不勾。
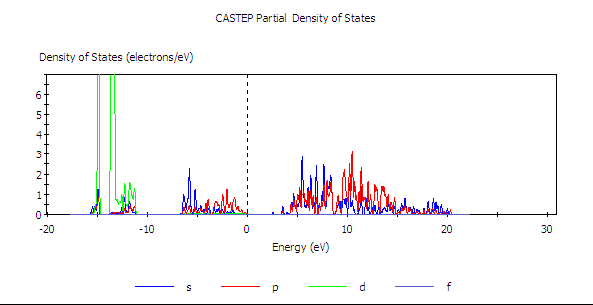
如此針對 Ga 與 N 所畫的兩張圖,在調整共同的尺度後,獲得以下兩張圖。
PDOS of Ga
PDOS of N
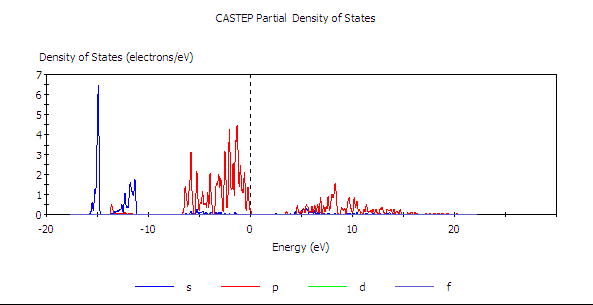
由結果可見,GaN 價帶的頂部是由氮所主導(而且是 p 軌域),導帶底部則是鎵與氧的混成,其中又以鎵貢獻得比較多。
二、
載入模型,改設成最小晶胞(Build -> Symmetry -> Primitive Cell)以節省時間。在 Task 選 Elastic Constants。由於是絕緣體,也可把 Electronic -> More -> SCF 中的 "fix occupancy" 勾起來節省計算時間(不勾也沒關係)。
算完後,在自動跳出的文字檔輸出或晶胞結構檔有打開的情況下,開啟分析表單,選 Elastic constants,之後按 Calculate,會得到另一文字檔,其中一段包含我們要的資訊(以 NaCl 為例):
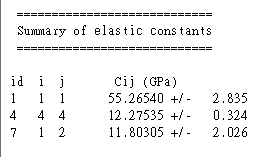
如果 Quality 是設成 Medium,並且 FFT 格子是 Standard 的話,會得到很不準確的結果,也就看不出什麼趨勢,如下:
|
|
C12
|
C44
|
|
LiF
|
24.5
|
63.1
|
|
NaCl
|
-6.9
|
12.3
|
|
MgO
|
71.8
|
130.1
|
反之,設定 Quality 至 Fine 並選用 FFT 格子密度為 Fine(該項目在 "Electronic -> More -> Basis" 下),則我們有更好的結果如下:
| |
C12
|
C44
|
|
LiF
|
39.9
|
61.9
|
|
NaCl
|
11.8
|
12.3
|
|
MgO
|
107.0
|
143.3
|
三、
After relax cell, one should do spin-polarised calcultion once again with cell fix (Task:Energy).
自發磁化率(單位 Bohr 磁子)
|
|
in cell
|
per atom
|
|
Ni3Cu
|
1.624
|
0.406
|
|
NiCu3
|
0.000
|
0.000
|
四、
載入碳的鑽石結構,轉成最小晶胞(以節省時間),Task 選 Energy 或 Geometry Optimisation 皆可,Properties 則加選 Optical Property,一般而言我們應針對總價電子帶的多寡來取未佔據態數(通常至少價帶數的兩倍以上),故預設之 12 個空軌域是不夠的,但這裏的價帶數是 4(每個碳四個價電子,晶胞內共兩個原子,故晶胞內共四個價電子帶), 因此預設值的 12 也就足夠了。
本題指定把能隙修正成為與實驗一致,因此我們需要知道未修正時能隙多少(至於實驗值則是題目己給),這時可利用光學計算的結果來作 TDOS 圖,如此可看出能隙(但記得把 smearing 定小一點,不要大過 0.1 eV,)
直接利用光學計算的結果也可以畫 TDOS (Total Density of States)
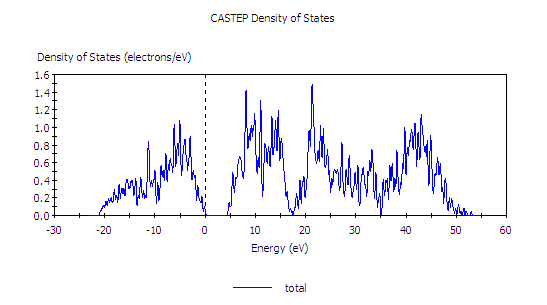
從上圖中,我們看出(必要時可用工具縮放尺度)與實驗值的 5.5 eV 尚差 1 eV 左右,因此可決定剪刀修正將採用 1.0 eV。
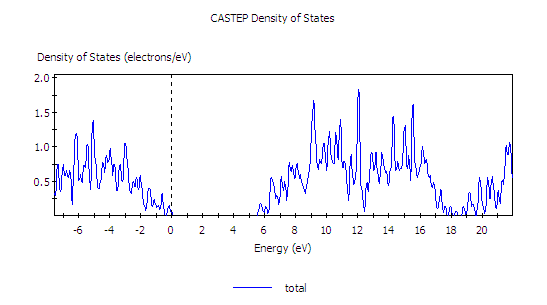
以下是在 smearing 0.05 eV 下,使用剪刀修正 1.0 eV 後 DOS 的呈現,已使能隙接近實驗值。
在前定的剪刀算符下(自動會設定成計算光學量要用的能隙 1.0 eV),如果不改預設的單位而使用 eV(光子能量),
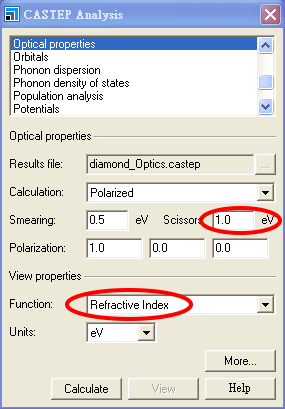
若在此時就按 Calculate 及之後的 View,則所畫出的譜線如下:
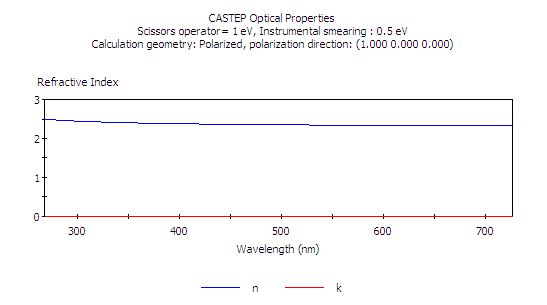
但本題所要求的,是呈現折射率隨波長的變化,因此我們應使用使用 nm 而非 eV 作為單位,
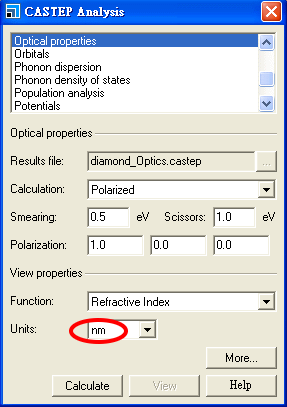
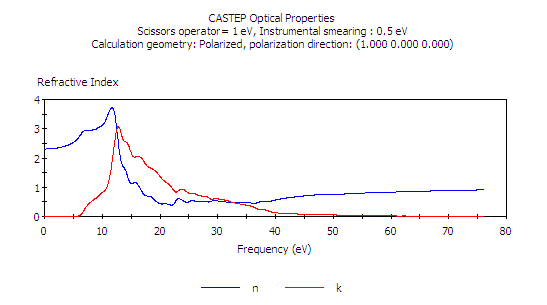
按 Calculate 及之後的 View,利用縮放及平移工具作調整

調整使其橫軸範圍涵蓋 300 nm 至 700 nm (即 3000 至 7000 埃),在可見光的波長範圍之內,我們會看到所計算的折射率符合實驗值。
五、
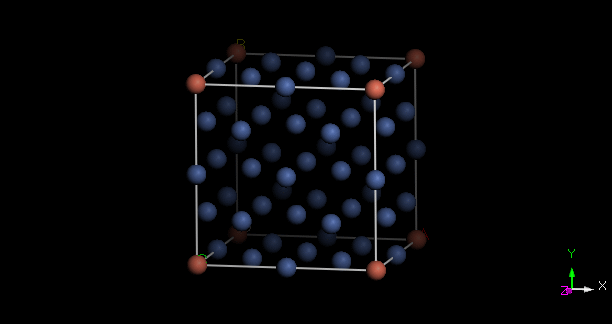
本題要求在 fcc 面心結構內做出最接近 3% 的摻雜,首先要思考最適合的超晶胞大小是什麼。面心結構在方方正正的傳統晶胞內有四個原子,若朝三個晶軸的方向各增加一倍長,則超晶胞體積變為 8 倍,有 32 顆原子。1/32 = 0.03125,已經很接近 0.03 了,因此我們在此做一個 Ni31Cu 的超晶胞。
載入 Ni 的傳統晶胞模型後,可使用 Build -> Symmetry -> Supercell 來做出它的 2x2x2 倍超晶胞,然中在其中任意取代一個原子為 Cu 即可。取代原子的作法如下:在模型中點選所要的原子,則 Property Explorer 的 Filter 會自動跳成 Atom,表列出該顆原子的所有性質:
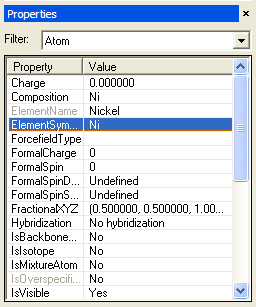
再點選 ElementSymbol 項目可逕自修改元素名稱。
雖說晶胞具有週期性邊界條件,選任何一個原子取代的效果皆相同,但為了分析操作或作圖顯示上的方便性,仍建議取此較 "明顯" 的位置,如中心或角落。
完成後超晶胞內原子眾多,不易觀察,可將滑鼠移至模型視窗內並按右鍵打開選單,進入 Display Options 功能表,將 Depth cue 強度調高
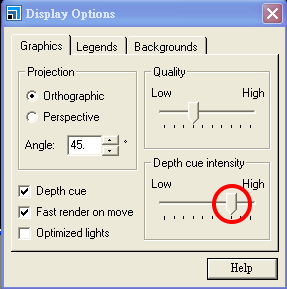
完成後之超晶胞結果如下:
取代的雜質原子置於中心

取代的雜子原子置於角落
若後續有針對結構進行計算的需要,提醒大家可到 Build -> Symmetry -> Find Symmetry 使用找對稱性工具並按 Impose 將該對稱性加諸在超晶胞之上,以節省計算。